Downloaded 153 times


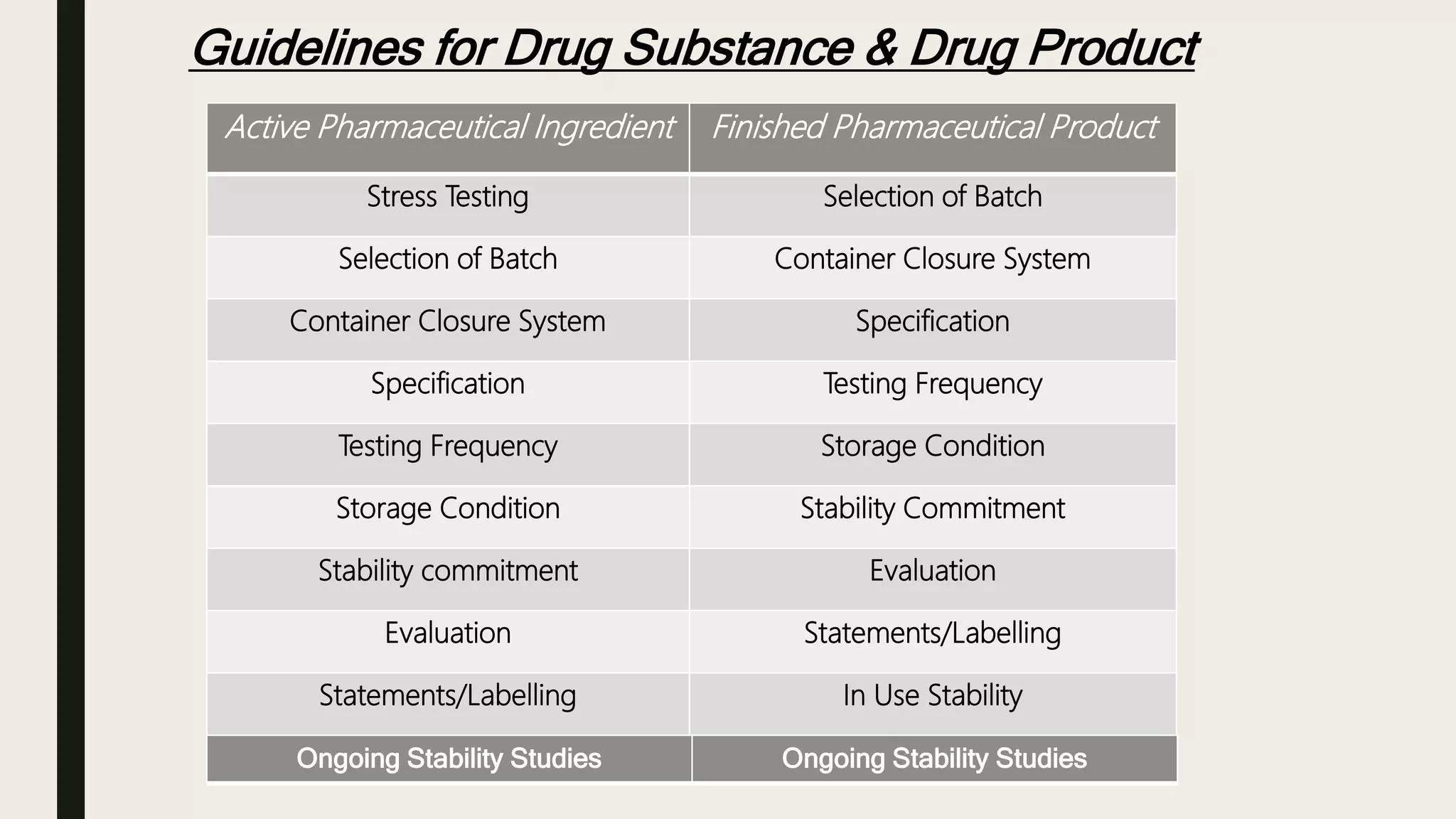


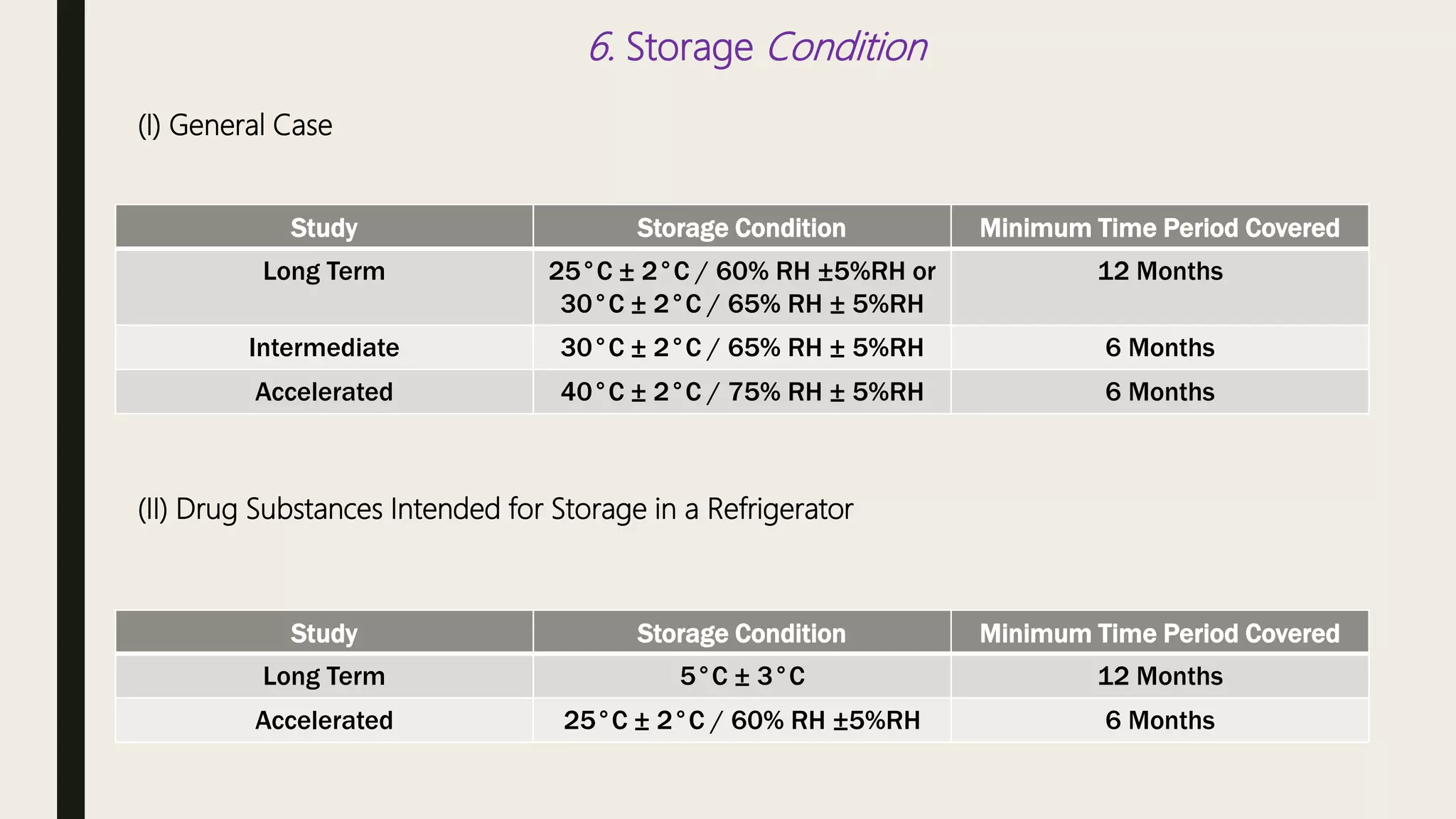
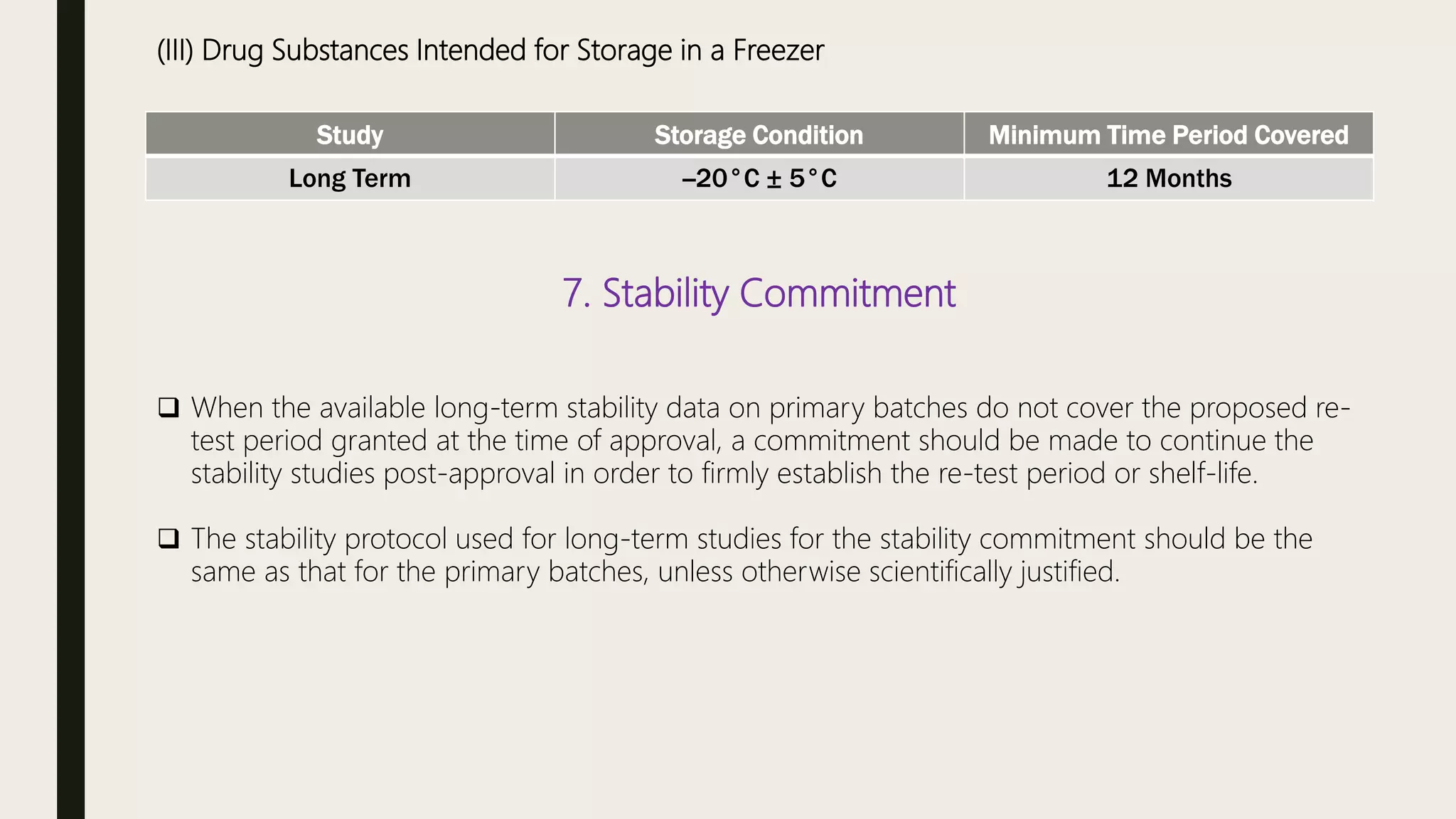



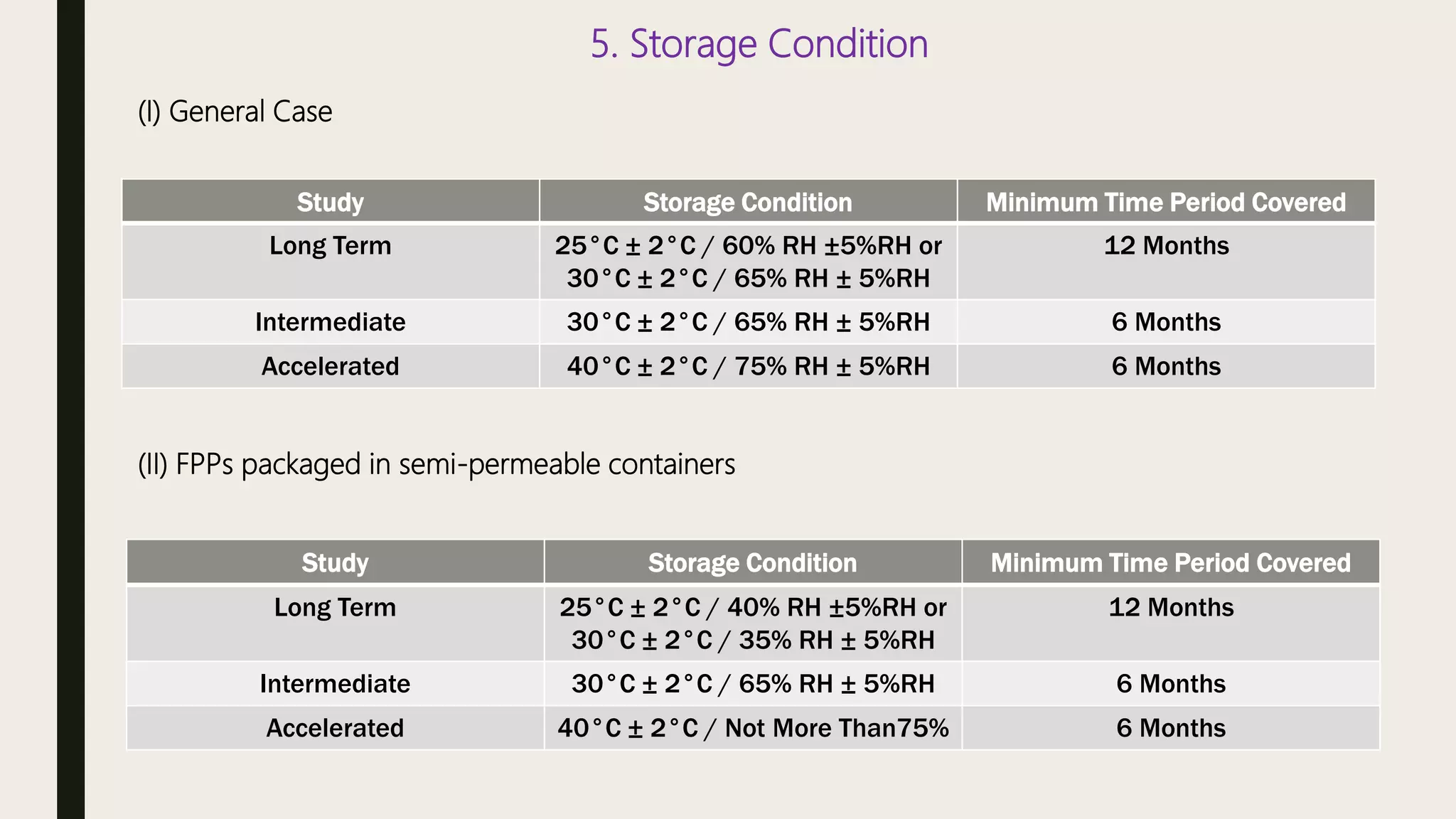
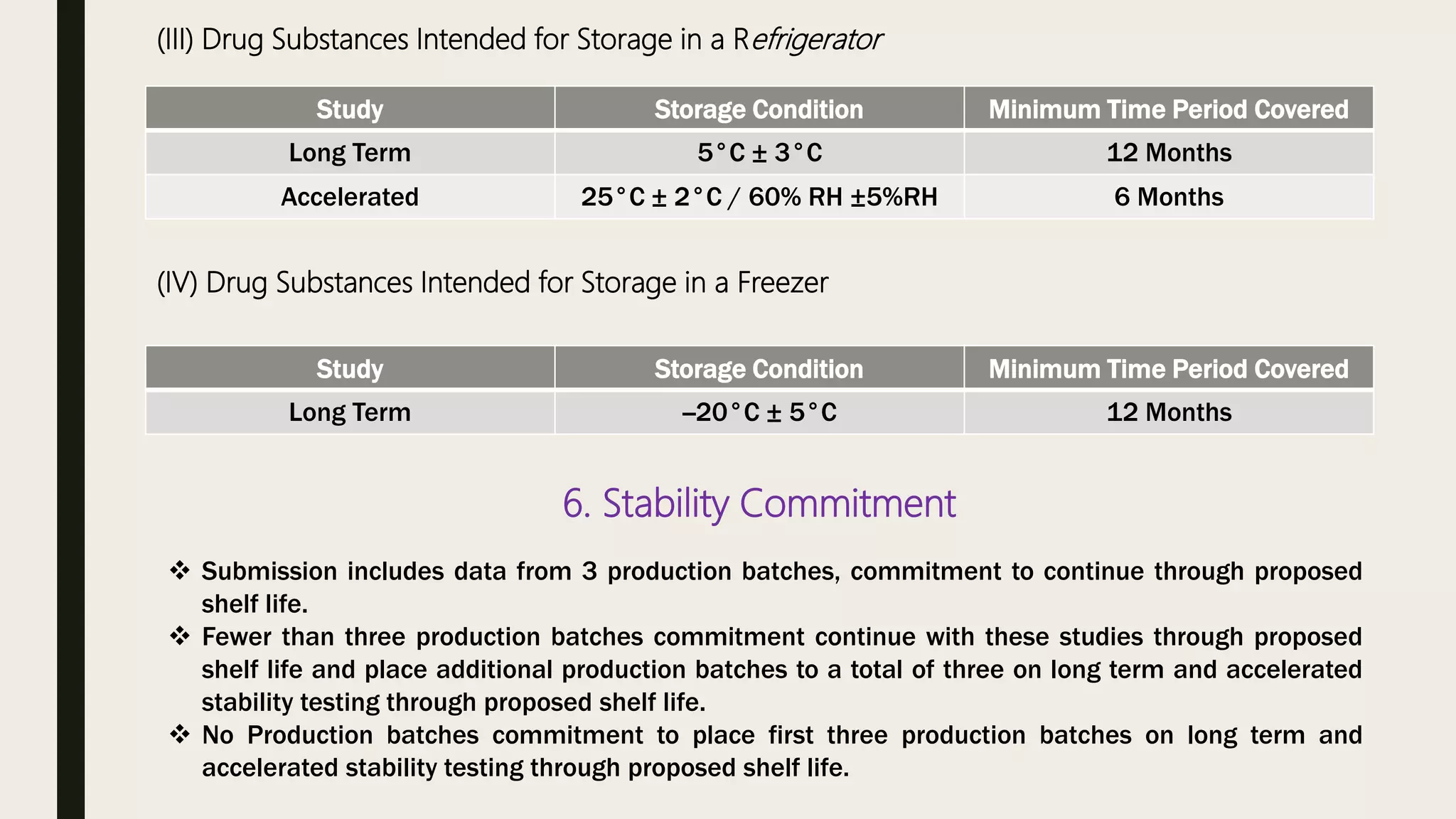


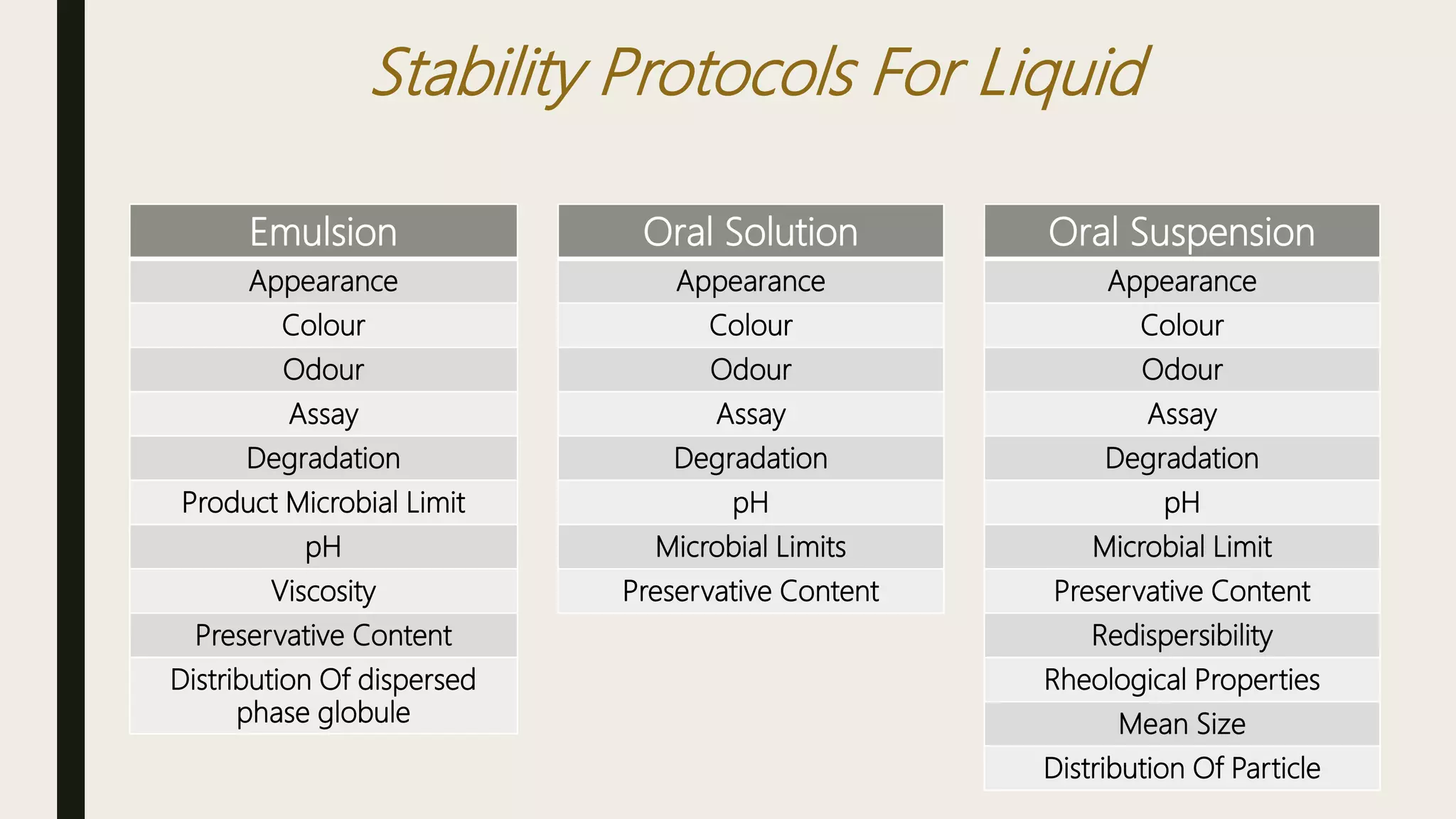
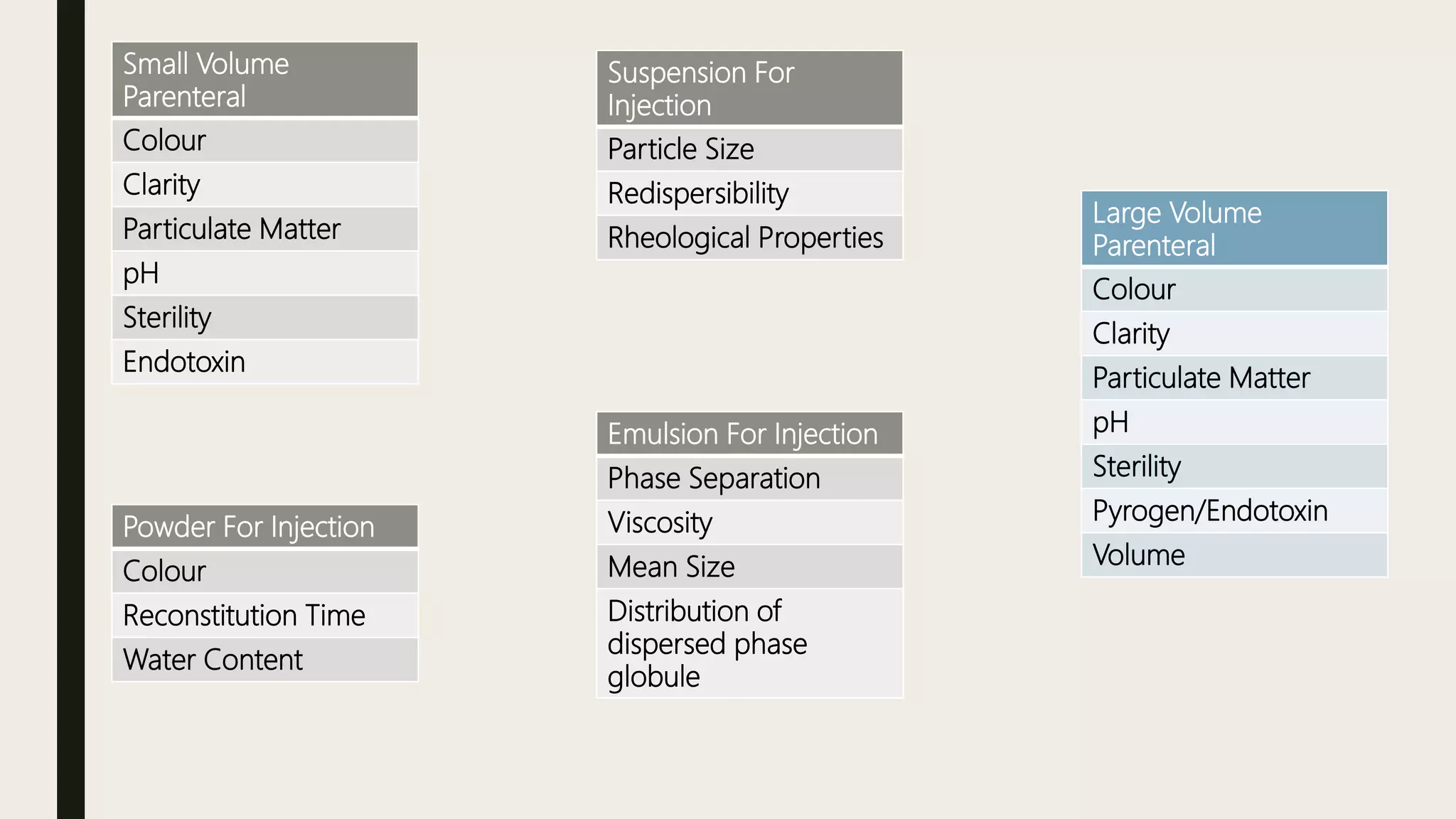





















This document summarizes WHO guidelines for stability testing of liquid dosage forms. It outlines the key aspects that should be covered in stability protocols, including specifications tested, storage conditions and minimum time periods for long term, intermediate and accelerated studies. Specific recommendations are provided for drug substances, oral solutions, suspensions, small volume parenterals and other dosage forms. The purpose of stability testing is to provide evidence of a product's quality over time under various environmental factors and establish a re-test period or shelf life. Ongoing stability studies are also required to monitor products throughout their shelf life.
























































































