Download to read offline



![FGCSForum
Roma,April24,2016
P..Misiser
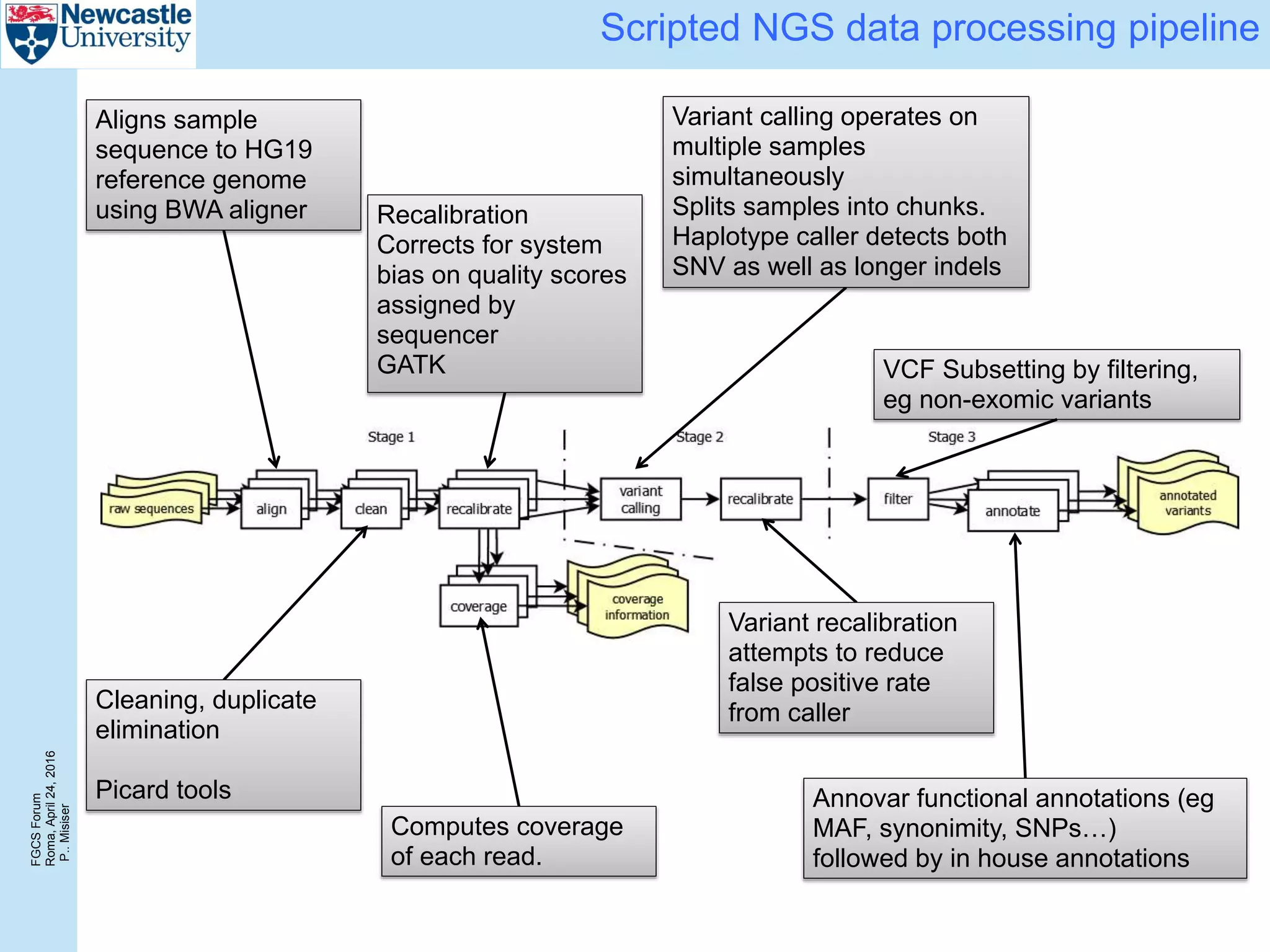
Performance
Configurations for 3VMs experiments:
HPC cluster (dedicated nodes):
3x8-core compute nodes Intel Xeon E5640, 2.67GHz CPU, 48 GiB RAM, 160
GB scratch space
Azure workflow engines:
D13 VMs with 8-core CPU, 56 GiB of memory and 400 GB SSD, Ubuntu 14.04.
00:00
12:00
24:00
36:00
48:00
60:00
72:00
0 6 12 18 24
Responsetime[hh:mm]
Number of samples
3 eng (24 cores) 6 eng (48 cores)
12 eng (96 cores)](https://image.slidesharecdn.com/2016-fgcs-forum-ceg-160424080508/75/Scalable-Whole-Exome-Sequence-Data-Processing-Using-Workflow-On-A-Cloud-16-2048.jpg)
![FGCSForum
Roma,April24,2016
P..Misiser
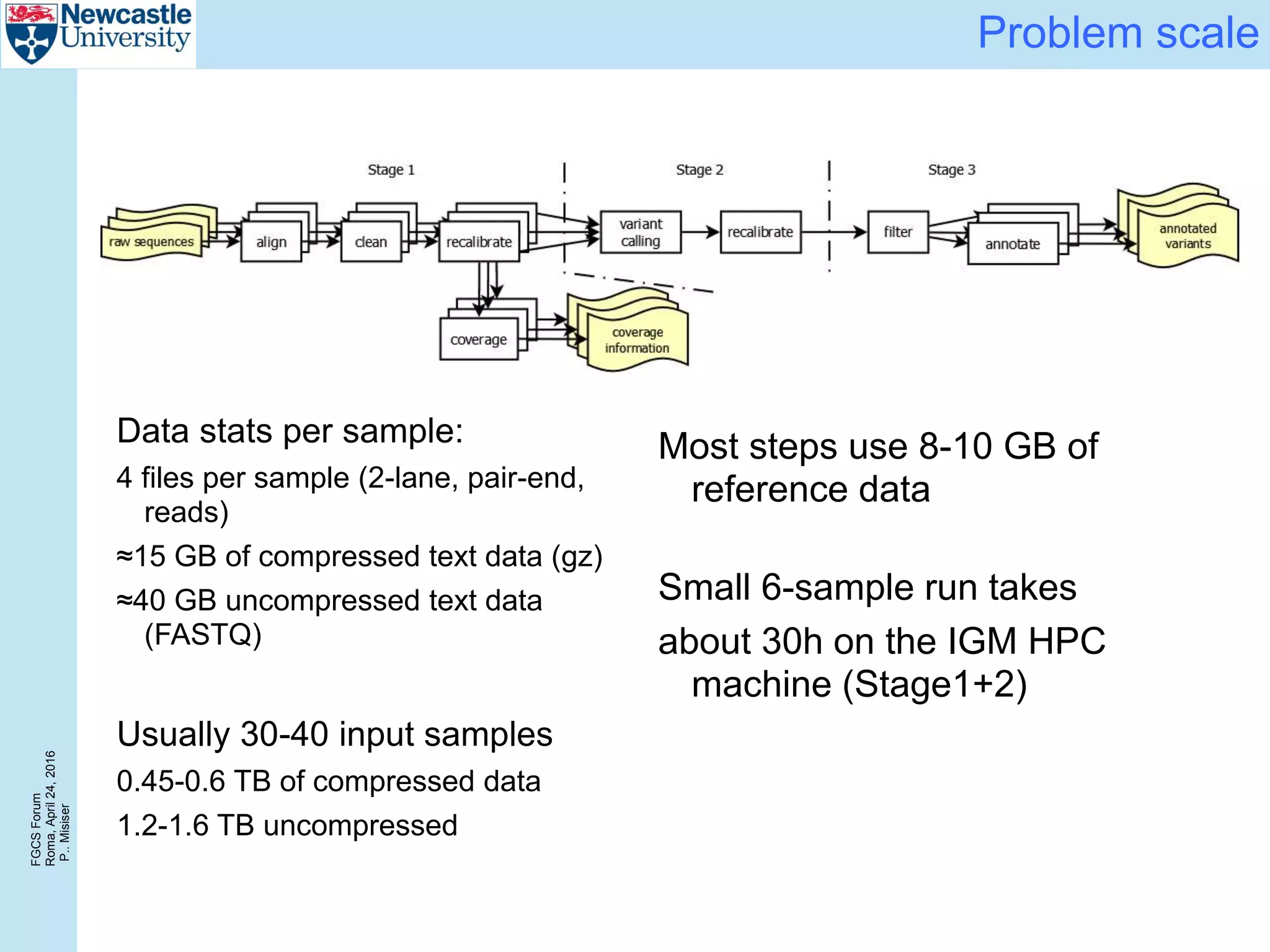
Comparison with HPC
0
24
48
72
96
120
144
168
0 6 12 18 24
Responsetime[hours]
Number of input samples
HPC (3 compute nodes) Azure (3xD13 – SSD) – sync Azure (3xD13 – SSD) – chained
0
1
2
3
4
5
6
0 50 100 150 200 250 300 350 400
Systemthroughput[GiB/hr]
Size of the sample cohort [GiB]
HPC (3 compute nodes) Azure (3xD13 – SSD) – sync Azure (3xD13 – SSD) – chained](https://image.slidesharecdn.com/2016-fgcs-forum-ceg-160424080508/75/Scalable-Whole-Exome-Sequence-Data-Processing-Using-Workflow-On-A-Cloud-17-2048.jpg)
![FGCSForum
Roma,April24,2016
P..Misiser
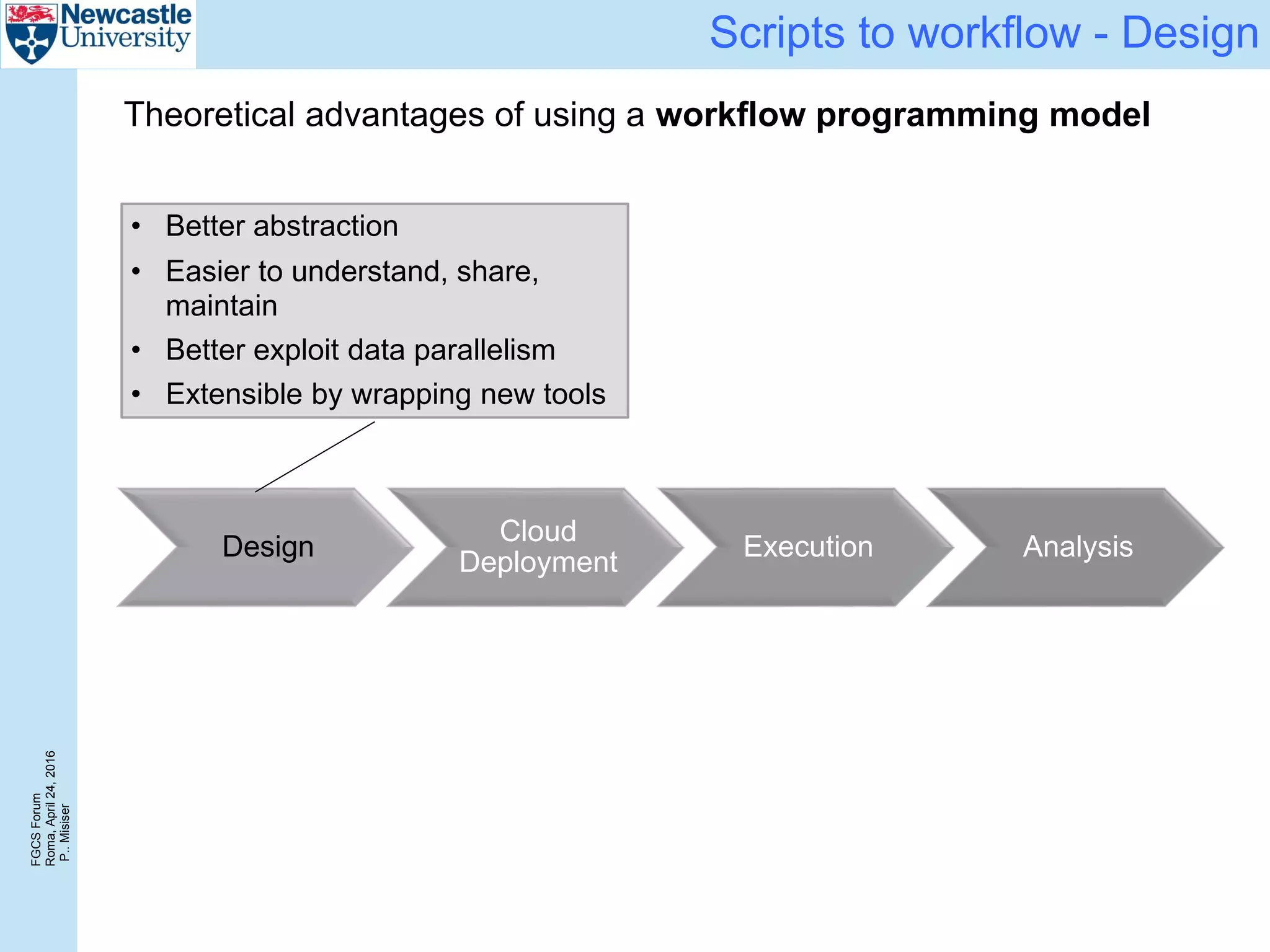
Cost
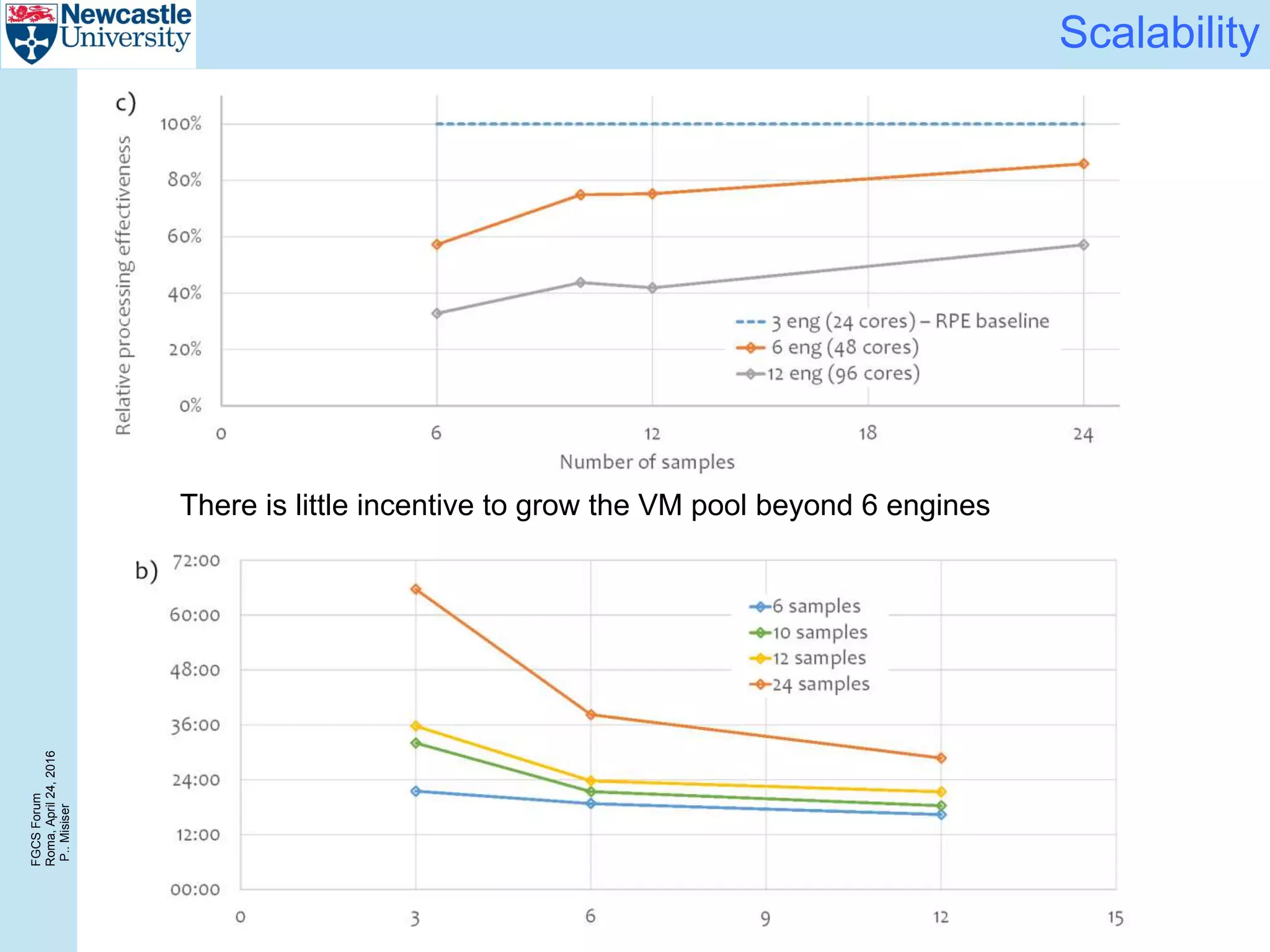
Again, a 6 engine configuration achieves near-optimal cost/sample
0 50 100 150 200 250 300 350
0
0.2
0.4
0.6
0.8
1
1.2
0 6 12 18 24
0
2
4
6
8
10
12
14
16
18
Size of the input data [GiB]
CostperGiB[£]
Number of samples
Costpersample[£]
3 eng (24 cores)
6 eng (48 cores)
12 eng (96 cores)](https://image.slidesharecdn.com/2016-fgcs-forum-ceg-160424080508/75/Scalable-Whole-Exome-Sequence-Data-Processing-Using-Workflow-On-A-Cloud-19-2048.jpg)















![FGCSForum
Roma,April24,2016
P..Misiser
Performance
Configurations for 3VMs experiments:
HPC cluster (dedicated nodes):
3x8-core compute nodes Intel Xeon E5640, 2.67GHz CPU, 48 GiB RAM, 160
GB scratch space
Azure workflow engines:
D13 VMs with 8-core CPU, 56 GiB of memory and 400 GB SSD, Ubuntu 14.04.
00:00
12:00
24:00
36:00
48:00
60:00
72:00
0 6 12 18 24
Responsetime[hh:mm]
Number of samples
3 eng (24 cores) 6 eng (48 cores)
12 eng (96 cores)](https://crownmelresort.com/image.slidesharecdn.com/2016-fgcs-forum-ceg-160424080508/75/Scalable-Whole-Exome-Sequence-Data-Processing-Using-Workflow-On-A-Cloud-16-2048.jpg)
![FGCSForum
Roma,April24,2016
P..Misiser
Comparison with HPC
0
24
48
72
96
120
144
168
0 6 12 18 24
Responsetime[hours]
Number of input samples
HPC (3 compute nodes) Azure (3xD13 – SSD) – sync Azure (3xD13 – SSD) – chained
0
1
2
3
4
5
6
0 50 100 150 200 250 300 350 400
Systemthroughput[GiB/hr]
Size of the sample cohort [GiB]
HPC (3 compute nodes) Azure (3xD13 – SSD) – sync Azure (3xD13 – SSD) – chained](https://crownmelresort.com/image.slidesharecdn.com/2016-fgcs-forum-ceg-160424080508/75/Scalable-Whole-Exome-Sequence-Data-Processing-Using-Workflow-On-A-Cloud-17-2048.jpg)

![FGCSForum
Roma,April24,2016
P..Misiser
Cost
Again, a 6 engine configuration achieves near-optimal cost/sample
0 50 100 150 200 250 300 350
0
0.2
0.4
0.6
0.8
1
1.2
0 6 12 18 24
0
2
4
6
8
10
12
14
16
18
Size of the input data [GiB]
CostperGiB[£]
Number of samples
Costpersample[£]
3 eng (24 cores)
6 eng (48 cores)
12 eng (96 cores)](https://crownmelresort.com/image.slidesharecdn.com/2016-fgcs-forum-ceg-160424080508/75/Scalable-Whole-Exome-Sequence-Data-Processing-Using-Workflow-On-A-Cloud-19-2048.jpg)


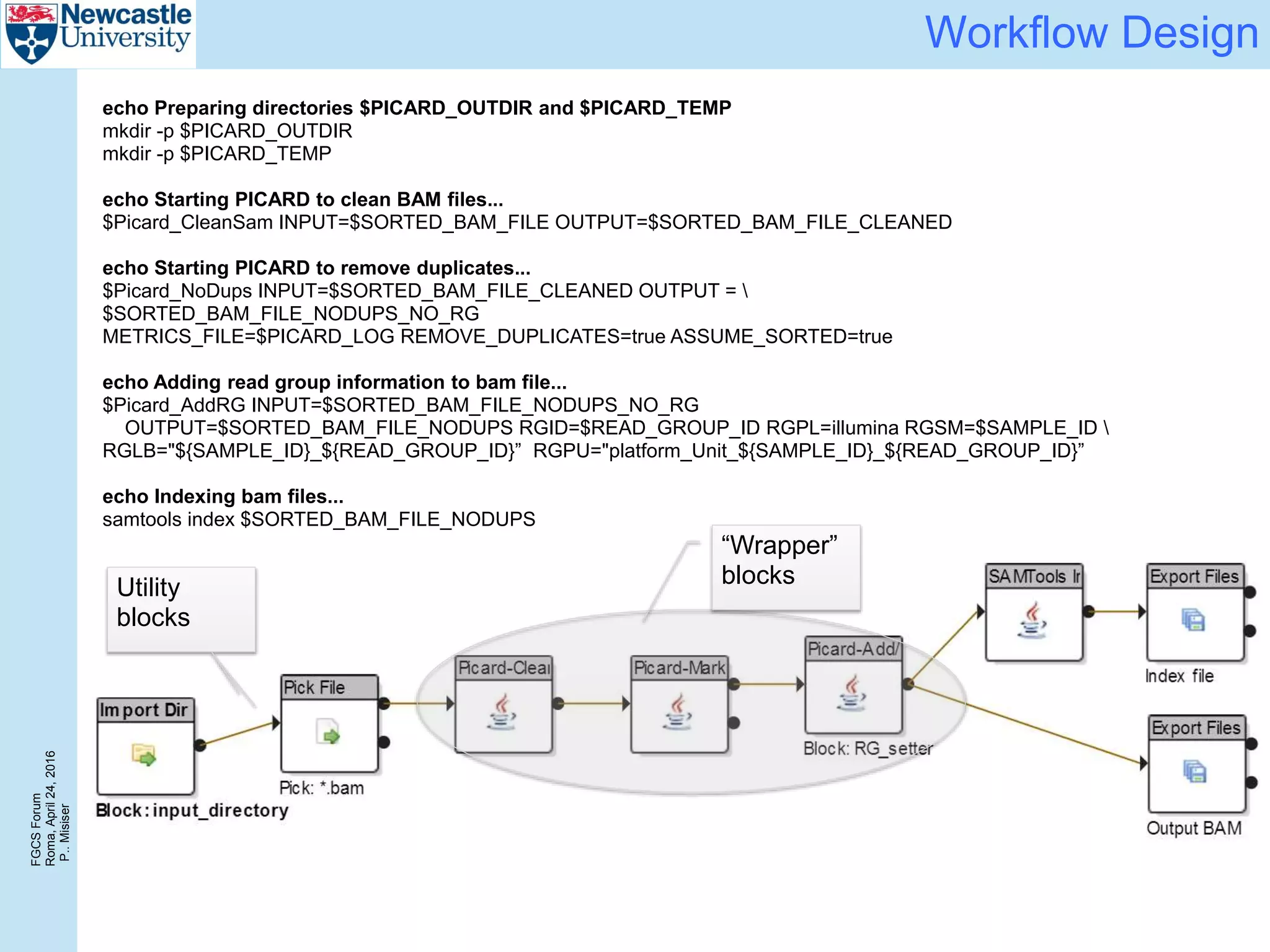
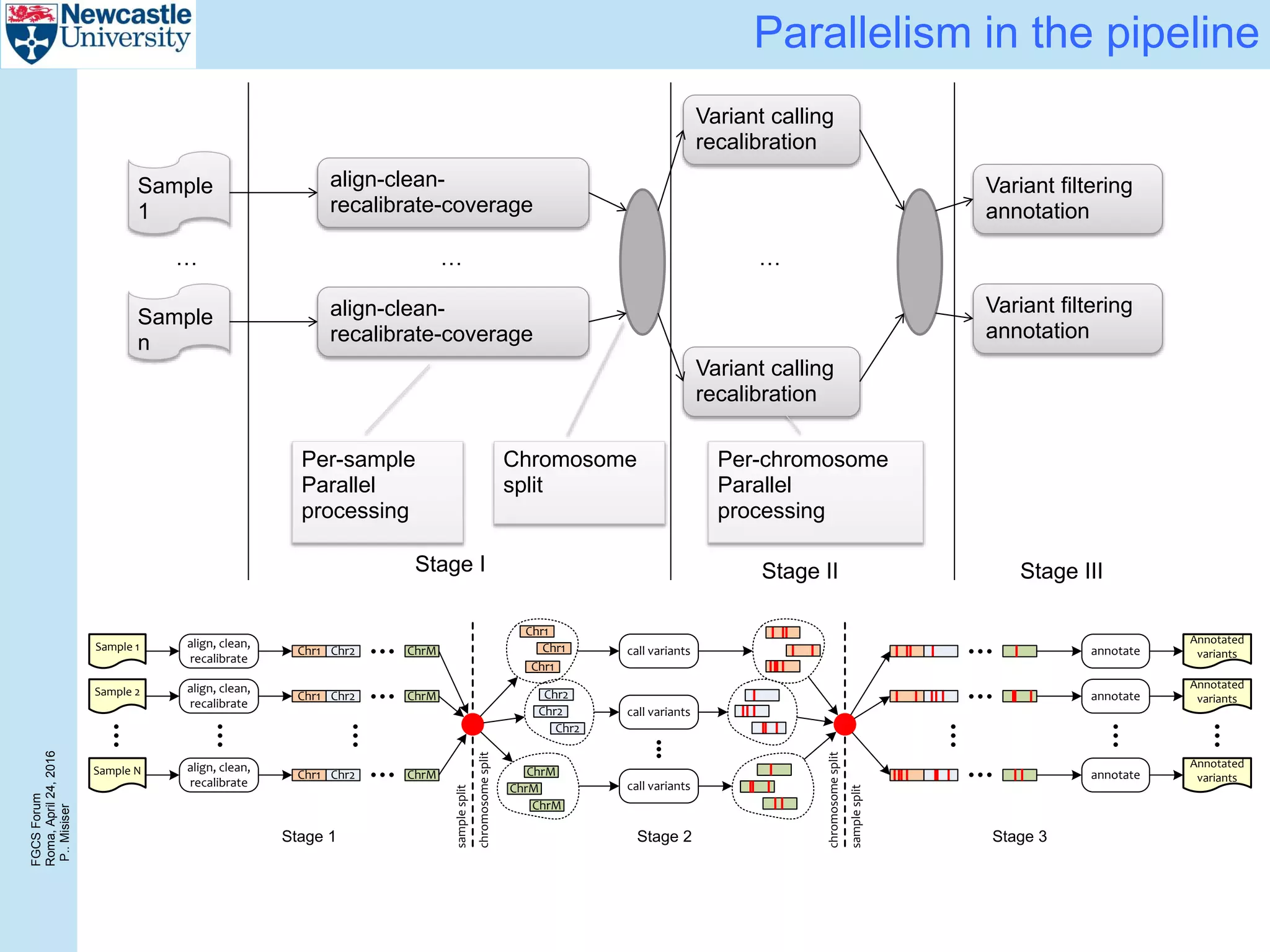
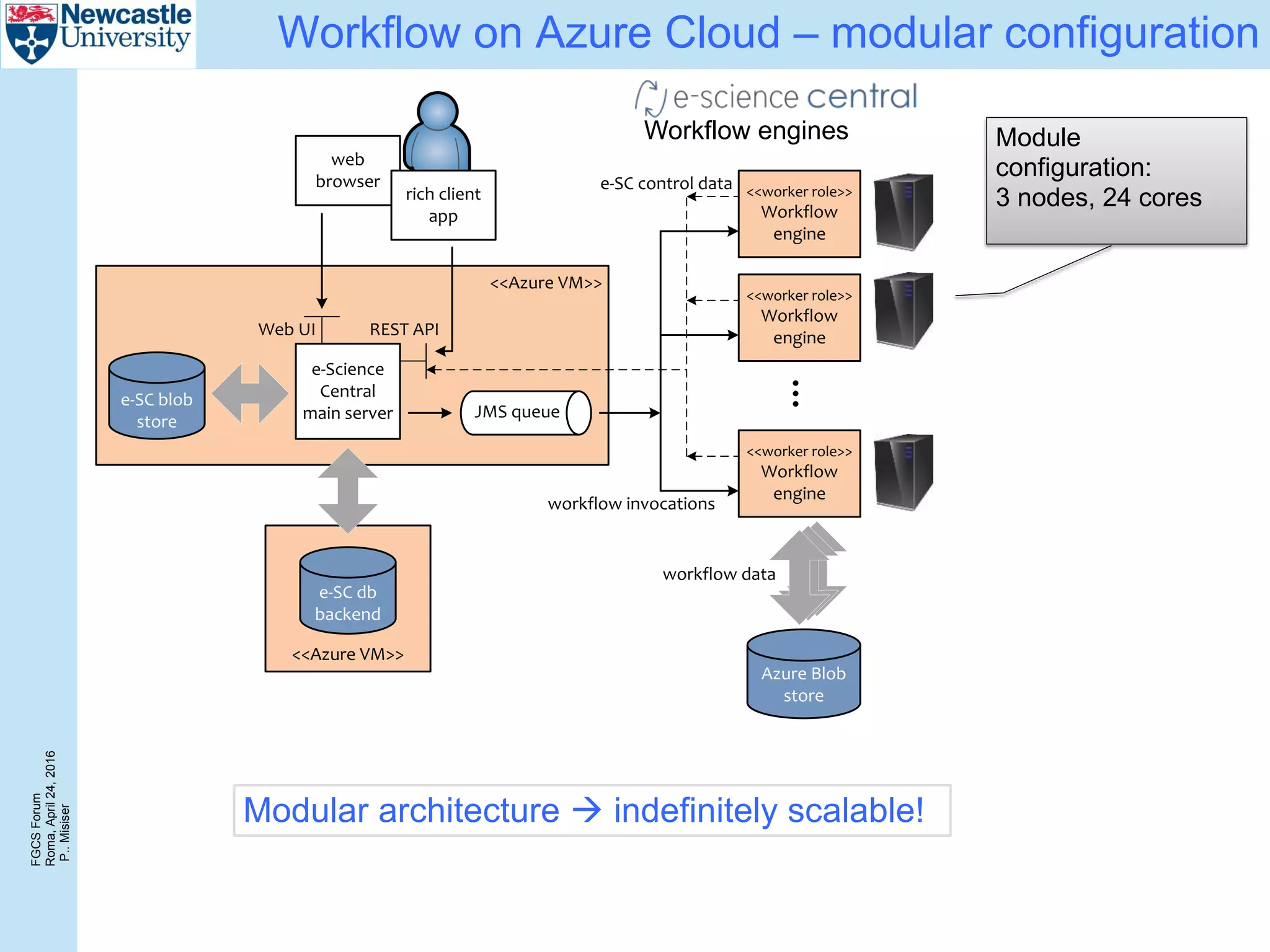
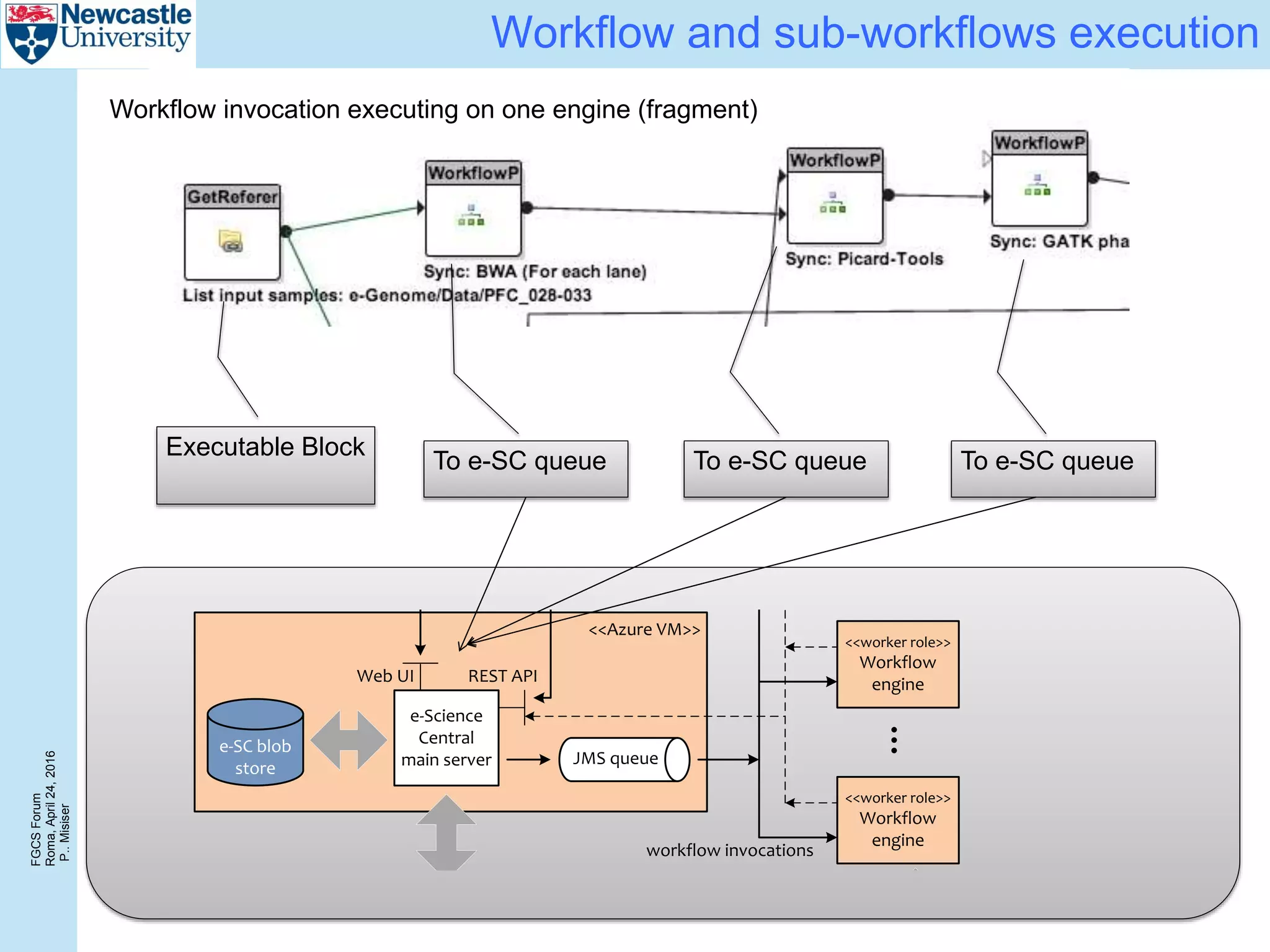
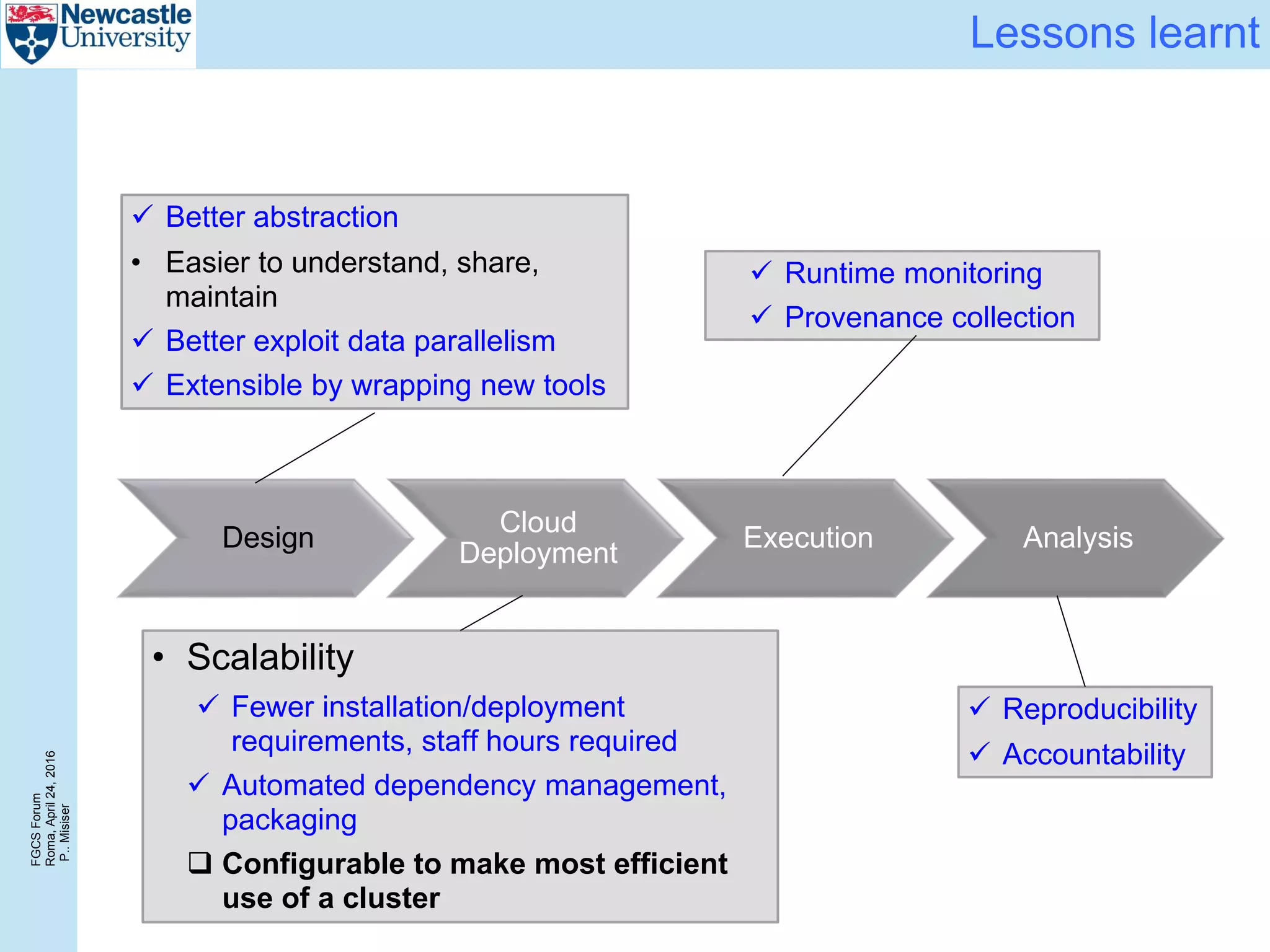
The document discusses the challenges and solutions in porting a whole-exome sequencing (WES) and whole-genome sequencing (WGS) pipeline from high-performance computing (HPC) to a public cloud, focusing on flexibility, performance, and abstraction. It outlines a scripted NGS data processing pipeline and evaluates its performance, scalability, and costs when using cloud resources, including Azure. Key benefits include better data parallelism, easier maintenance and sharing of workflows, and automating dependency management.


















































































